
The above painting depicts electron microscopy of two glomerular capillary loops and a mesangial region from a case of membranous lupus nephritis, showing intramembranous and mesangial electron-dense deposits. An electron photomicrograph from a patient with membranous lupus nephritis, showing the same features, is also shown below.
Pure membranous lupus nephritis (classified under the International Society of Nephrology / Renal Pathology Society system as class V), without an associated focal or diffuse (proliferative) lupus nephritis, occurs in approximately 15 percent of all patients with renal involvement by systemic lupus erythematosus (SLE). This percentage is higher in African Americans with SLE. While focal or diffuse lupus nephritis tends to occur in patients with positive serologic studies (including anti-dsDNA antibodies) and hypocomplementemia, membranous lupus nephritis can occur in patients without anti-dsDNA antibodies and who have normal serum complement levels. Patients with membranous lupus nephritis present with proteinuria, which can be sub-nephrotic or nephrotic range, and often have microscopic hematuria. Hypertension is also common. Patients with membranous lupus nephritis, as with patients with idiopathic membranous glomerulopathy, also have an increased risk of thrombotic complications associated with nephrotic range proteinuria. These include renal vein thrombosis, deep venous thromboses, and pulmonary emboli.
Features distinguishing membranous lupus nephritis from idiopathic membranous glomerulopathy include negativity for the phospholipase A2 receptor (PLA2R) and thrombospondin type 1 domain-containing 7a (THSD7A) antigens, presence of mesangial and/or subendothelial electron-dense deposits, subendothelial tubuloreticular structures, presence of a tissue ANA pattern, “full house” immunofluorescence (with IgA, IgG, IgM, C3, and C1q deposition within glomeruli), a lack of IgG4 subclass specificity, and positivity for exostosin (EXT). These distinguishing features are not seen in all cases. Some cases of membranous lupus nephritis lack a “full house” immunofluorescence pattern and express only IgG, C3, and light chains. The subset of cases that are EXT-associated may also be IgG4 subclass restricted (Sethi et al, 2019). In addition, subendothelial tubuloreticular structures or a tissue ANA pattern are not seen within all cases.
Membranous lupus nephritis has a variable prognosis, with some patients having an indolent course, and approximately 10% progressing to end-stage renal disease (Mercadal et al, 2002). Disease progression can occur due to increased interstitial fibrosis due to damage from persistent nephrotic range proteinuria, or by transition to a proliferative form of lupus nephritis. Factors that predict disease progression, in cases lacking a concurrent proliferative lupus nephritis, are currently unclear and is an important area of investigation. Treatment is indicated for patients with severe or persistent nephrotic syndrome, an increased serum creatinine, or who have an active urinary sediment (concerning for a concurrent proliferative component).
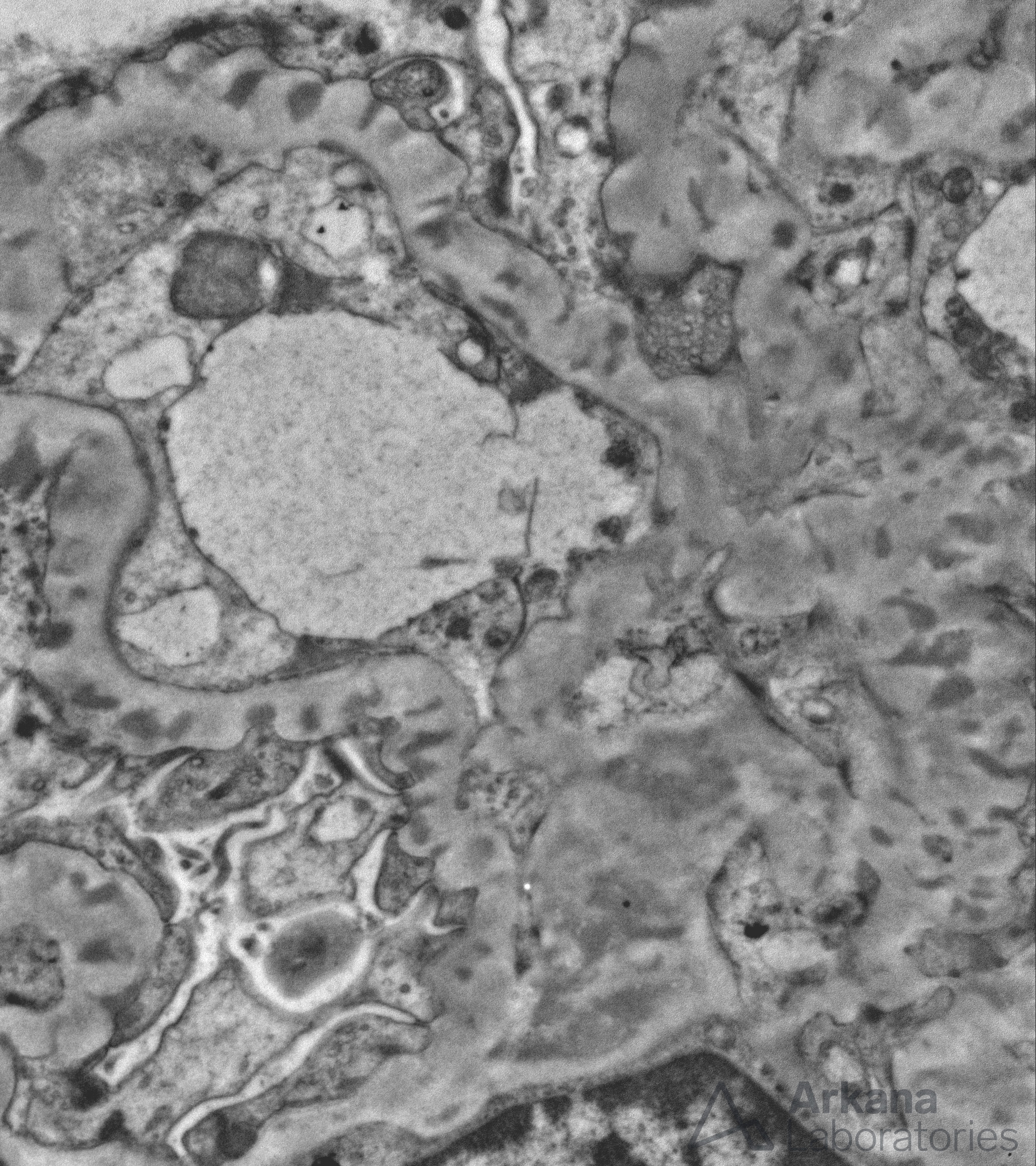
Electron photomicrograph showing subepithelial, intramembranous, and mesangial electron-dense deposits within a glomerulus from a patient with membranous lupus nephritis.
References:
Mercadal L, Montcel ST, Nochy D, Queffeulou G, Piette JC, Isnard-Bagnis C, Martinez F. Factors affecting outcome and prognosis in membranous lupus nephropathy. Nephrol Dial Transplant 2002; 17: 1771-1778.
Sethi S, Madden BJ, Debiec H, Charlesworth MC, Gross L, Ravindran A, Hummel AM, Specks U, Fervenza FC, Ronco P. Exostosin 1/Exostosin-2-Associated Membranous Nephropathy. J Am Soc Nephrol 2019 Jun; 30 (6): 1123-1136.
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.
