
The above painting depicts electron microscopy of a case of Alport syndrome. Alport syndrome is a type of hereditary nephritis due to mutations in the alpha 3, alpha 4, or alpha 5 chains of collagen type IV. As a result, the typical alpha 3-4-5 meshwork of type IV collagen in the GBM is replaced by an alpha 1-2 meshwork, which has less tensile strength and has susceptibility to proteolysis by matrix metalloproteinases (Zeisberg et al, 2006).
Patients with Alport syndrome present with hematuria and progressive renal failure, and males are disproportionately affected. Deafness and visual problems can also occur. Kidney biopsy findings can be suggestive, but not completely diagnostic, of Alport syndrome, which is confirmed by genetic testing by next-generation sequencing. Kidney biopsy findings by light microscopy include focal segmental glomerulosclerosis, interstitial foam cells, and interstitial fibrosis and tubular atrophy. Mesangial hypercellularity is present in some cases. Electron microscopy is crucial in these cases to examine for abnormalities of the glomerular basement membranes (Adam et al, 2013).
Ultrastructural findings that are most characteristic of Alport syndrome are multilamination and thinning of the glomerular basement membranes. Multilamination increases with age, and results in segmental thickening of the GBM. There are small densities, but not true electron dense immune-type deposits that are between the layers (“bread crumbs”). Scalloping occurs both externally and internally along the GBM at the subepithelial and subendothelial surface. Scalloping refers to outpouching that occurs with GBM remodeling. Podocyte foot process effacement can be extensive and may be associated with microvillous transformation. The abnormal GBMs and podocyte foot process effacement can result in high grade proteinuria.
Here are some ultrastructural images of Alport syndrome from a patient’s case.
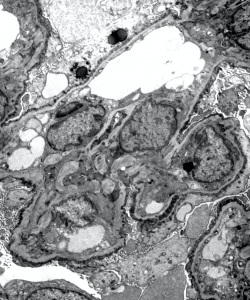
1) The above EM image shows segmentally thinned and thickened areas of glomerular basement membrane. There is prominent scalloping seen along the GBMs and podocyte foot process effacement.
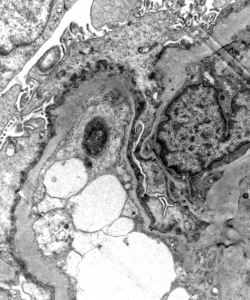
2) This image shows a glomerular capillary loop with prominent external scalloping, podocyte foot process effacement, and microvillous transformation.
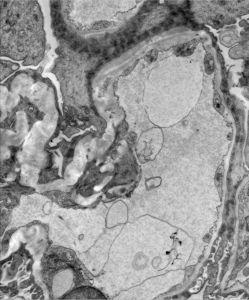
3) Splitting / lamination “basket weaving” along the gglomerular basement membrane is shown in the image above.
When these findings are seen by electron microscopy, genetic testing should be considered, especially in young male patients or with a positive family history of kidney disease. The differential diagnosis for ultrastructural GBM abnormalities includes Fechtner syndrome (characterized by GBM abnormalities, deafness, and cataracts), Epstein syndrome (characterized by GBM abnormalities, enlarged platelets, thrombocytopenia, and basophilic inclusions within WBC), Denys-Drash syndrome (GBM abnormalities at a very young age <2 years, can progress to diffuse mesangial sclerosis and ESKD in early childhood), and other familial forms of focal segmental glomerulosclerosis. A remote immune complex-mediated glomerulonephritis can also result in ultrastructural GBM abnormalities due to resolution and remodeling of the GBM, but do not commonly have all of these constellation of findings.
References:
Adam J, Connor TMF, Wood K, Lewis D, Naik R, Gale DP, Sayer JA. Genetic testing can resolve diagnostic confusion in Alport syndrome. Clinical Kidney Journal 2014 April; 7 (2): 197-200.
Colvin, Chang (Eds). Diagnostic Pathology: Kidney Diseases Third Edition (2019), Elsevier Publishing, pp: 352-355.
Zeisberg M, Khurana M, Rao VH, Cosgrove D, Rougier JP, Werner MC, Shield CF 3rd, Werb Z, Kalluri R. Stage-specific action of matrix metalloproteinases influences hereditary kidney disease. PLoS Medicine 2006; 3: 535-546.
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.
Share this article:
Related Posts
