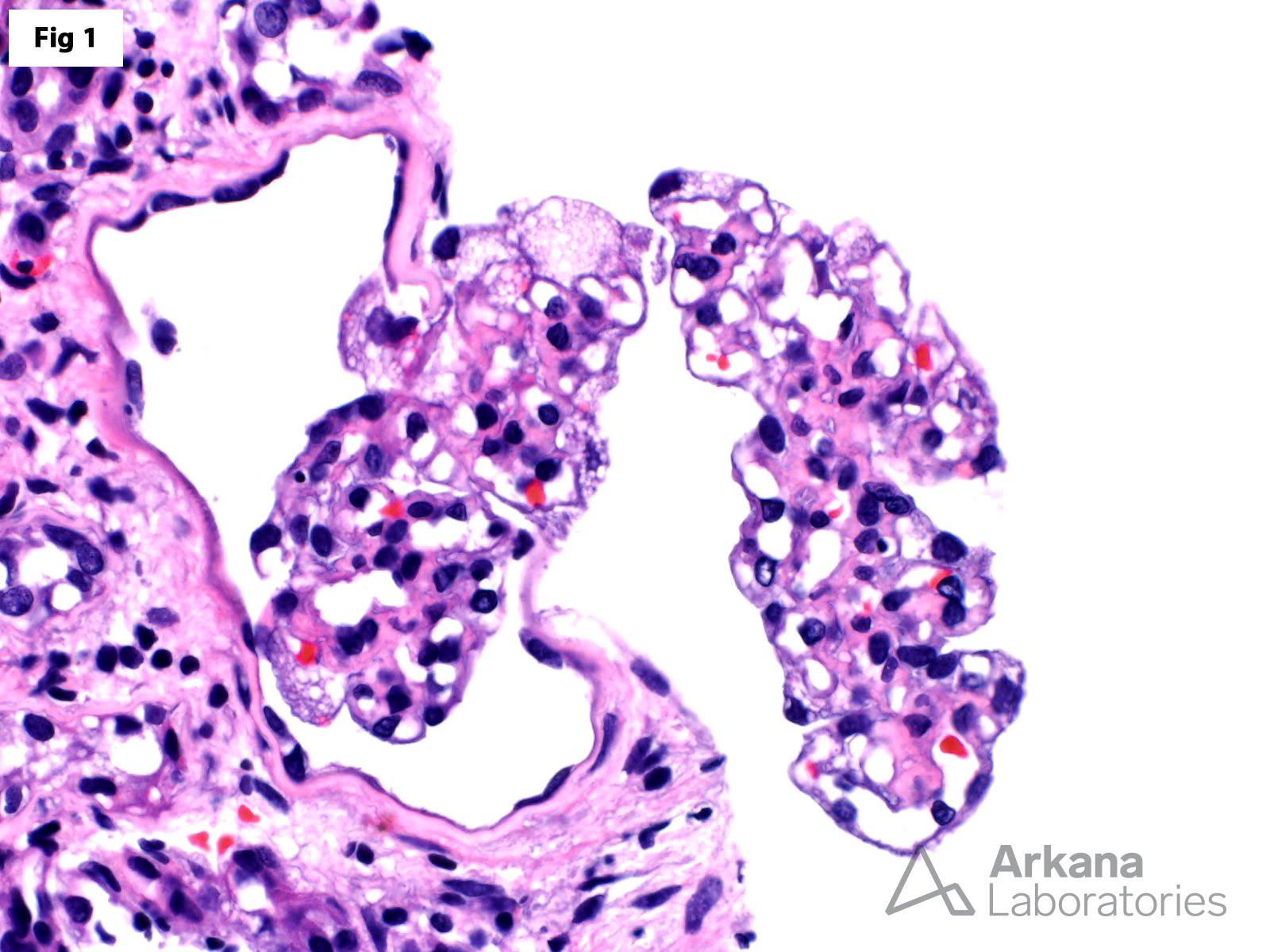
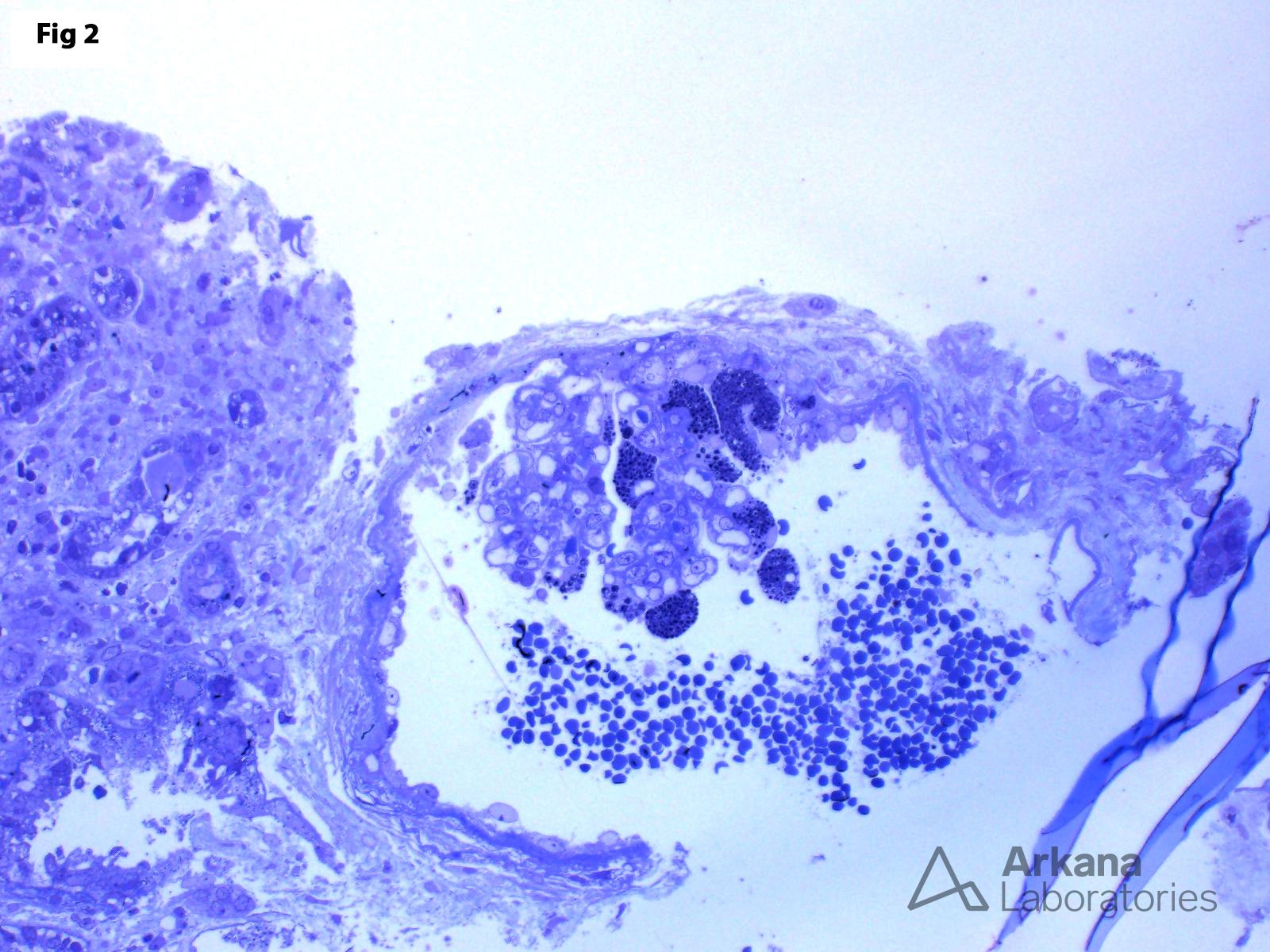
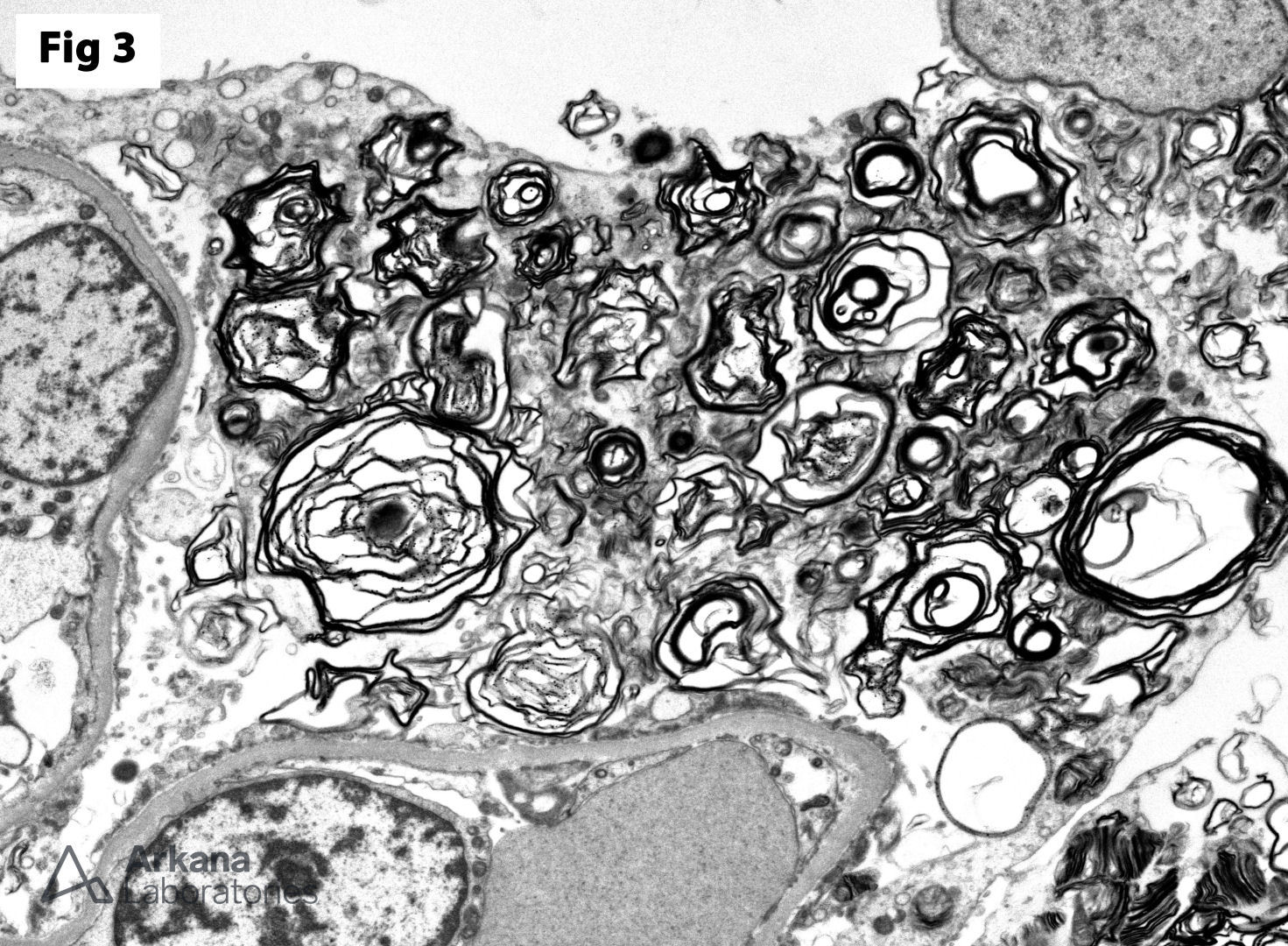
This biopsy shows glomeruli with visceral epithelial cells (podocytes) displaying ample vacuolated cytoplasm (Fig 1), which is highlighted on a toluidine blue stain (Fig 2). More importantly, the electron microscopy shows numerous large, lamellated lipid vacuoles (myeloid bodies) within the podocytes, which are characteristic of Fabry disease (Fig 3). Fabry disease is an X-linked recessive disorder caused by a deficiency of the lysosomal enzyme α-galactosidase A, which catalyzes the cleavage of glycosphingolipids. Deficiency of this enzyme results in lysosomal accumulation of globotriaosylceramide in the heart, kidney, eye, nerves, sweat glands and vascular endothelium. Specifically, within the kidney, globotriaosylceramide may accumulate within endothelial cells, podocytes, distal tubular cells and collecting duct epithelium. The clinical presentation mostly depends on the residual enzymatic activity. The most severe end of the spectrum is characterized by renal dysfunction which progresses to end-stage renal disease by 40 years of age, which is accompanied by hypertrophic cardiomyopathy, acroparesthesias, angiokeratomas, anhidrosis and corneal opacities. On the other hand, milder enzyme deficiencies may present as cardiac or renal variants without showing the full spectrum of the disorder. The renal biopsy is particularly helpful in the diagnosis of this disorder. Identification of myeloid bodies by electron microscopy is highly suggestive, but not diagnostic of the disease, given that similar inclusions may be seen in patients treated with amiodarone, chloroquine, and hydroxychloroquine.
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.